Une maladie respiratoire rare pourrait être plus répandue au Québec
Une équipe de chercheurs a découvert une forme génétique rare de dyskinésie ciliaire primitive liée à un seul ancêtre ayant vécu il y a plus de 300 ans, laissant présager l’existence de cas plus légers et non diagnostiqués dans le Bas-Saint-Laurent et la Côte-du-Sud.
Une étude menée à l’Institut de recherche du Centre universitaire de santé McGill (L’Institut) a identifié un variant génétique rare du gène ODAD4 responsable de la dyskinésie ciliaire primitive (DCP), une maladie héréditaire chronique qui affecte le système respiratoire. Grâce à des analyses génétiques et généalogiques approfondies, l’équipe de recherche a trouvé des preuves solides indiquant que ce variant remonterait probablement à un ancêtre unique ayant vécu il y a environ 330 ans, et qu’il pourrait expliquer des cas non détectés de DCP à travers le Québec — en particulier dans les régions du Bas-Saint-Laurent et de la Côte-du-Sud.
L’étude révèle également que ce variant peut mener à une forme plus légère et jusqu’ici non reconnue de la maladie. Les conclusions des chercheurs, publiées dans la revue scientifique Cells, pourraient expliquer pourquoi certaines personnes non diagnostiquées souffrent de sinusite chronique, d’otites récurrentes, d’une toux grasse persistante ou d’une congestion nasale sévère depuis la naissance.
La DCP est une maladie génétique rare qui altère la structure et la fonction des cils, de minuscules structures en forme de poils qui aident à évacuer le mucus des voies respiratoires. Lorsque les cils ne fonctionnent pas correctement, le mucus s’accumule, entraînant des infections chroniques et une clairance mucociliaire inefficace. Avec le temps, les personnes atteintes de DCP développent généralement des bronchectasies, un élargissement irréversible des bronches visible à l’imagerie médicale et provoquant une accumulation persistante de mucus.

« Cette étude est la première à décrire une forme plus légère de la maladie qui entraîne des symptômes chroniques des voies respiratoires supérieures, mais sans nécessairement causer de lésions pulmonaires visibles, et, surtout, à démontrer qu’elle est très probablement associée à un variant fondateur québécois », explique le Dr Adam Shapiro, auteur principal de l’étude et fondateur de la clinique de DCP du Centre universitaire de santé McGill (CUSM), la seule clinique spécialisée en DCP au Québec offrant des tests diagnostiques de pointe, incluant des analyses du phénotype et du génotype.
« Nos résultats ont des implications cliniques immédiates. Ils signifient qu’il y a des personnes au Québec, particulièrement dans le Bas-Saint-Laurent et la Côte-du-Sud, qui sont probablement atteintes de DCP, mais qui n’ont jamais été évaluées, car leurs poumons semblent en “trop bon état” », ajoute-t-il.
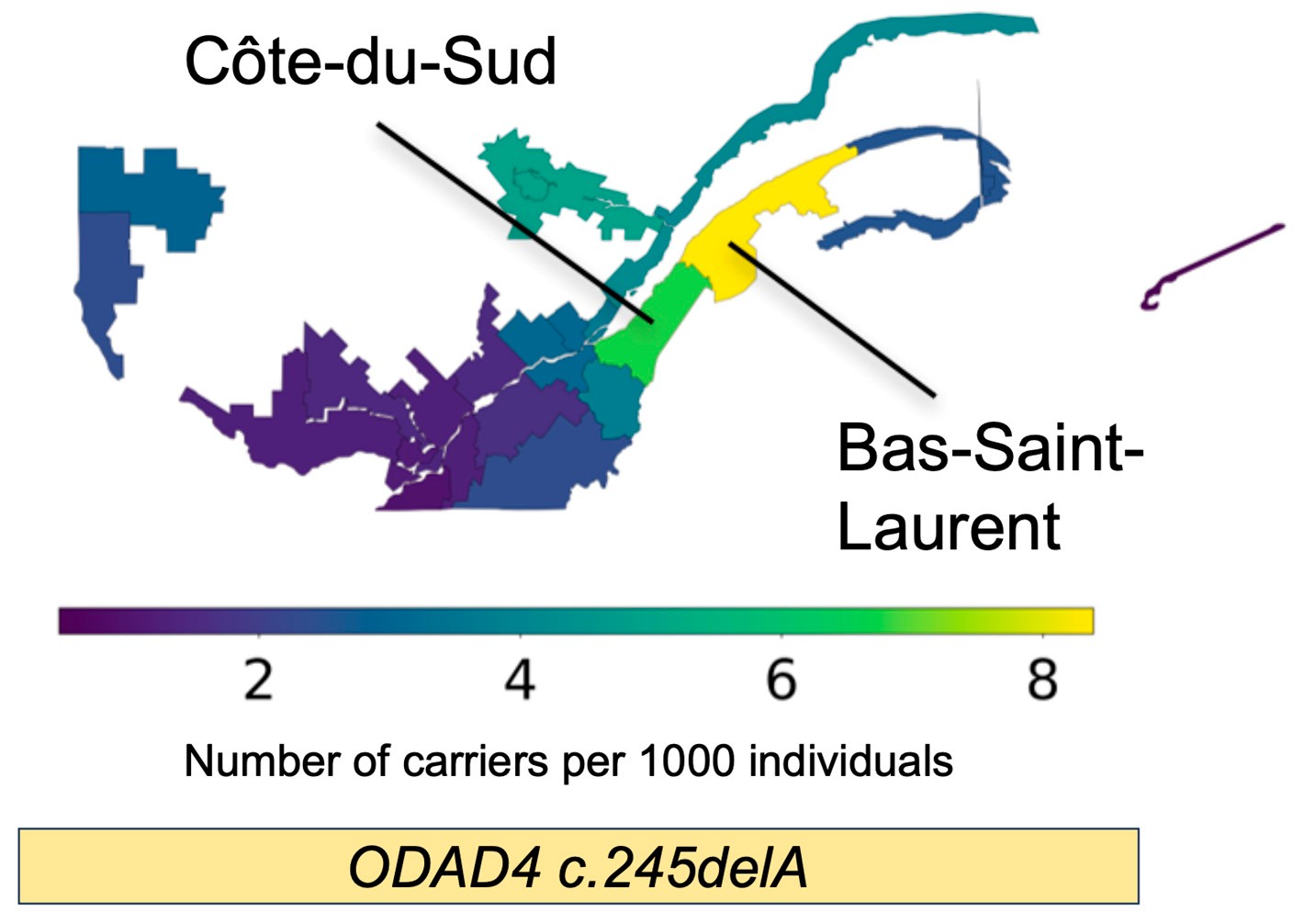
Les analyses des chercheurs ont révélé que les porteurs du nouveau variant c.245delA descendent probablement d’un ancêtre unique ayant vécu à l’époque de la colonisation française du Québec. Des simulations ont également montré que le variant c.245delA est plus fréquent dans deux régions de l’est du Québec situées sur la rive sud du fleuve Saint-Laurent. Dans ces régions, la fréquence estimée du variant est la suivante :
- Bas-Saint-Laurent: 1 personne sur 111
- Côte-du-Sud: 1 personne sur 143
« Cette forte concentration géographique suggère un effet fondateur évident et indique que d’autres cas de DCP pourraient exister dans ces régions. Cela signifie que des médecins pourraient, sans le savoir, traiter des patients dont les problèmes respiratoires sont causés par le variant c.245delA », explique le Dr Shapiro, également chercheur au sein du Programme de recherche en santé de l’enfant et en développement humain de L’Institut, pneumologue pédiatrique à l’Hôpital de Montréal pour enfants du CUSM et professeur agrégé au Département de pédiatrie de l’Université McGill.
Reproduit de Bourassa et al. (2025), « ODAD4-related primary ciliary dyskinesia: report of five cases and a founder variant in Quebec », Cells, 14(18), 1460. https://doi.org/10.3390/cells14181460. CC BY 4.0.
De la découverte de cinq patients à une découverte populationnelle : comment un variant fondateur québécois a émergé
Dans cette étude, l’équipe de recherche a décrit les cas de cinq individus sans lien familial et atteints de DCP causée par des variants bialléliques du gène ODAD4, c’est-à-dire des variants dans lesquels les deux copies du gène, l’une héritée de chaque parent, comportent une altération pathogène. Ces personnes ont été identifiées grâce à des références cliniques à la clinique de DCP du CUSM et par l’intermédiaire du Genetic Disorders of Mucociliary Clearance Consortium (GDMCC), un réseau collaboratif regroupant huit sites de recherche en Amérique du Nord et dirigé par l’Université de Caroline du Nord (UNC). Des chercheurs de l’Université McGill, de l’UNC et de l’Université Yale ont également contribué à ces travaux.
Trois de ces individus étaient d’ascendance canadienne-française et présentaient le même nouveau variant homozygote, c.245delA, tandis que les deux autres cas, identifiés aux États-Unis, portaient d’autres variants du gène ODAD4.
Fait remarquable, deux patients québécois porteurs du variant c.245delA, âgés de 14 et 38 ans, présentaient des symptômes des voies respiratoires inférieures inhabituellement légers, sans signe de bronchectasies. Malgré cette atteinte pulmonaire plus modérée, les deux individus souffraient néanmoins d’une sinusite chronique importante, d’otites récurrentes et d’une toux grasse persistante. L’un des patients présentait également des problèmes de fertilité, une autre conséquence fréquente de la DCP, en raison du rôle essentiel des cils dans le déplacement des spermatozoïdes et des ovules.
« La raison pour laquelle le variant c.245delA dans le gène ODAD4 est associé à une forme plus bénigne de la maladie chez certaines personnes reste incertaine. Le dépistage régional de ce variant au Québec pourrait aider à identifier d’autres cas et à approfondir notre compréhension de cette forme nouvellement découverte de DCP », explique le Dr Shapiro.
Pour déterminer si les cas québécois partageaient une origine commune, les chercheurs ont combiné le séquençage complet du génome, des outils de modélisation génétique de pointe et quatre siècles de données généalogiques. À l’aide de la base de données populationnelles CARTaGENE, qui regroupe des données génétiques et de santé provenant de 43 000 Québécois, ils ont pu repérer des porteurs du variant rare d’ODAD4. Des outils analytiques avancés ont ensuite permis à l’équipe de retracer la transmission du variant au fil des générations et de relier, avec leur consentement, les porteurs identifiés à la base de données BALSAC, qui rassemble plus de 400 ans de registres de naissances, de mariages et de décès provenant de l’ensemble du Québec.
La prochaine étape : accroître les diagnostics grâce au dépistage génétique ciblé
Lorsque la DCP est diagnostiquée tôt, un traitement peut être offert pour en ralentir la progression. Malheureusement, comme les symptômes de la DCP ressemblent à ceux de nombreuses affections respiratoires beaucoup plus courantes, la maladie passe souvent inaperçue. Le diagnostic est également complexe : il nécessite plusieurs évaluations et un équipement spécialisé pour exclure d’autres maladies, ainsi que des tests génétiques permettant d’identifier les variants responsables du trouble.
« Nos résultats mettent en évidence l’importance du dépistage génétique ciblé et d’une collaboration renforcée avec les médecins des régions où ce variant est plus répandu, explique le Dr Shapiro. Nous invitons les cliniciens de ces régions à orienter les patients présentant des symptômes respiratoires chroniques inexpliqués vers une évaluation pour la DCP. Cela nous permettra non seulement de poser un diagnostic précis et d’offrir des soins appropriés, mais aussi de mieux comprendre le comportement de ce variant rare dans la population et de mettre au jour les mécanismes qui sous-tendent les formes plus légères de DCP. Au final, ces connaissances contribueront au développement de traitements plus efficaces. »
À propos de l’étude
“ODAD4-related primary ciliary dyskinesia: report of five cases and a founder variant in Quebec” ” a été rédigé par Marie-Hélène Bourassa, Guillaume Sillon, Shuizi Ding, Maurizio Chioccioli, Monkol Lek, Kaiyue Ma, Alejandro Mejia-Garcia, Simon Gravel, Donald C. Vinh, Michael R. Knowles, MargaretW. Leigh, Stephanie D. Davis, Thomas Ferkol, Kenneth N. Olivier, Elizabeth N. Schecterman, Weining Yin, Patrick R. Sears, Martina Gentzsch, Susan E. Boyles, William D. Bennett, Kirby L. Zeman, Lawrence E. Ostrowski, Maimoona A. Zariwala et Adam J. Shapiro, et publié dans Cells.
DOI: https://doi.org/10.3390/cells14181460
Les chercheurs remercient les personnes atteintes de DCP et leurs familles pour leur participation.
Ces travaux de recherche ont été financés par le Département de pédiatrie de l’Université McGill et la Fondation de l’Hôpital de Montréal pour enfants, et soutenus directement ou indirectement par divers organismes, notamment le Genetic Disorders of Mucociliary Clearance Consortium, le National Center for Advancing Translational Sciences, le National Heart, Lung, and Blood Institute, l’American Thoracic Society Foundation, Génome Québec (Ministère de l’Économie, de l’Innovation et de l’Énergie), Partenariat canadien contre le cancer et Santé Canada.
Personne-ressource pour les médias
Fabienne Landry
Coordonnatrice des communications, Recherche, CUSM
[email protected]